

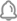


















































































































U19国青1-0击败越南队后久尔杰维奇:赢下比赛很重要
6月5日,四国赛U19国青1-0战胜越南U19。赛后,U19国青主帅久尔杰维奇接受了采访,对球队的表现进行了评价。 久尔杰维奇首先恭喜了越南队的出色表现,并表示这是U19国青本次四国赛的首场比赛,也是
 球会友谊 球会友谊
球会友谊 球会友谊  格鲁杯 格鲁杯 格鲁杯
格鲁杯 格鲁杯 格鲁杯  巴圣青联
巴圣青联  德堡州联
德堡州联  巴青锦标 球会友谊 球会友谊 巴青锦标 巴青锦标 巴青锦标
巴青锦标 球会友谊 球会友谊 巴青锦标 巴青锦标 巴青锦标  斯伐杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊
斯伐杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊  哥伦U19 球会友谊
哥伦U19 球会友谊  巴马全联 巴马全联 巴马全联 斯伐杯 斯伐杯
巴马全联 巴马全联 巴马全联 斯伐杯 斯伐杯  墨西女U19
墨西女U19  斯伐甲 球会友谊 球会友谊 球会友谊 球会友谊 斯伐杯
斯伐甲 球会友谊 球会友谊 球会友谊 球会友谊 斯伐杯  匈丙 球会友谊 球会友谊 球会友谊
匈丙 球会友谊 球会友谊 球会友谊  秘后备 球会友谊 秘后备
秘后备 球会友谊 秘后备  塞内联 格鲁杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 格鲁杯 格鲁杯 格鲁杯 巴圣青联 德堡州联 巴青锦标 球会友谊 球会友谊 巴青锦标 巴青锦标 巴青锦标 斯伐杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 哥伦U19 球会友谊 巴马全联 巴马全联 巴马全联 斯伐杯 斯伐杯 墨西女U19 斯伐甲 球会友谊 球会友谊 球会友谊 球会友谊 斯伐杯 匈丙 球会友谊 球会友谊 球会友谊 秘后备 球会友谊 秘后备 格鲁杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊
塞内联 格鲁杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 格鲁杯 格鲁杯 格鲁杯 巴圣青联 德堡州联 巴青锦标 球会友谊 球会友谊 巴青锦标 巴青锦标 巴青锦标 斯伐杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 哥伦U19 球会友谊 巴马全联 巴马全联 巴马全联 斯伐杯 斯伐杯 墨西女U19 斯伐甲 球会友谊 球会友谊 球会友谊 球会友谊 斯伐杯 匈丙 球会友谊 球会友谊 球会友谊 秘后备 球会友谊 秘后备 格鲁杯 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊 球会友谊  危地乙
危地乙 热点新闻
Hot NEWS
6月5日,四国赛U19国青1-0战胜越南U19。赛后,U19国青主帅久尔杰维奇接受了采访,对球队的表现进行了评价。 久尔杰维奇首先恭喜了越南队的出色表现,并表示这是U19国青本次四国赛的首场比赛,也是
 收藏(988)
收藏(988)  阅读(988)
阅读(988) 
6月4日,中国女足在一场友谊赛中以0比2不敌澳大利亚女足。赛后,中国女足前锋王霜在社交媒体上发文,表达了对此次澳洲之旅的看法,并晒出了自己的球衣以及全队的大合照。王霜在社交媒体上写道:“继
收藏(286) 阅读(286) 
6月6日,近日,中国男足将在主场迎战泰国队,历史数据统计显示,中国队在与泰国队的交锋中占据着较大的优势。两队共交手30场比赛,中国队取得20胜4平6负的战绩,打进66球,仅丢24球。其中,1988年2
收藏(217) 阅读(217) 
6月6日,中国男足将在沈阳主场迎战泰国队,这将是双方第31次交锋。据统计,国足在之前的30次交锋中取得了20场胜利,并打进了66球。射手榜公布后,宿茂臻以5个进球排名第一,其中1个是帽子戏法,成为国足
收藏(688) 阅读(688) 精彩视频
Wonderful video热门球队
FAVOURITE| 排名 | 球队名 | 总场次 |
1 |
2 |
|
2 |
1 |
|
3 |
3 |
|
4 |
1 |
|
5 |
1 |
|
6 |
2 |
|
7 |
1 |
|
8 |
1 |
|
9 |
6 |
|
10 |
2 |
|
11 |
2 |
|
12 |
1 |
|
13 |
4 |
|
14 |
2 |
|
15 |
1 |
| 排名 | 球队名 | 总场次 |
1 |
9 |
|
2 |
6 |
|
3 |
15 |
|
4 |
6 |
|
5 |
3 |
|
6 |
2 |
|
7 |
7 |
|
8 |
4 |
|
9 |
1 |
|
10 |
4 |
|
11 |
2 |
|
12 |
2 |
|
13 |
2 |
|
14 |
4 |
|
15 |
2 |
| 排名 | 球队名 | 总场次 |
1 |
1 |
|
2 |
1 |
|
3 |
1 |
|
4 |
2 |
|
5 |
1 |
|
6 |
1 |
|
7 |
1 |
|
8 |
1 |
|
9 |
2 |
|
10 |
2 |
| 排名 | 球队名 | 总场次 |
1 |
3 |
|
2 |
2 |
|
3 |
1 |
|
4 |
2 |
|
5 |
1 |
|
6 |
1 |
|
7 |
4 |
|
8 |
1 |
|
9 |
2 |
|
10 |
1 |
|
11 |
1 |
|
12 |
1 |
|
13 |
1 |
|
14 |
1 |
|
15 |
1 |
| 排名 | 球队名 | 总场次 |
1 |
1 |
|
2 |
2 |
|
3 |
1 |
|
4 |
1 |
|
5 |
1 |
|
6 |
1 |
|
7 |
0 |
|
8 |
1 |
|
9 |
1 |
|
10 |
2 |
|
11 |
1 |
|
12 |
1 |
|
13 |
1 |
|
14 |
1 |
|
15 |
1 |
| 排名 | 球队名 | 总场次 |
1 |
1 |
|
2 |
0 |
|
3 |
1 |
|
4 |
1 |
|
5 |
1 |
|
6 |
1 |
|
7 |
1 |
|
8 |
1 |
|
9 |
1 |
|
10 |
2 |
|
11 |
1 |
|
12 |
1 |
|
13 |
2 |
|
14 |
1 |
|
15 |
1 |

















































































































